DriverMap™ Genome-Wide Expression Profiling Solution
Complete Quantitative Gene Expression Analysis of All Human Genes in a One-Tube Assay
DriverMap™ Genome-Wide Expression Profiling Solution
- 100-fold more sensitive than RNA-Seq-detect 20-30% more low-abundance transcripts
- Start with as little as 10 pg total RNA-single-cell level
- Use total RNA from whole blood or tissues-no mRNA enrichment or globin-depletion
- Specific targeted primers-minimal background from mouse when analyzing xenograft
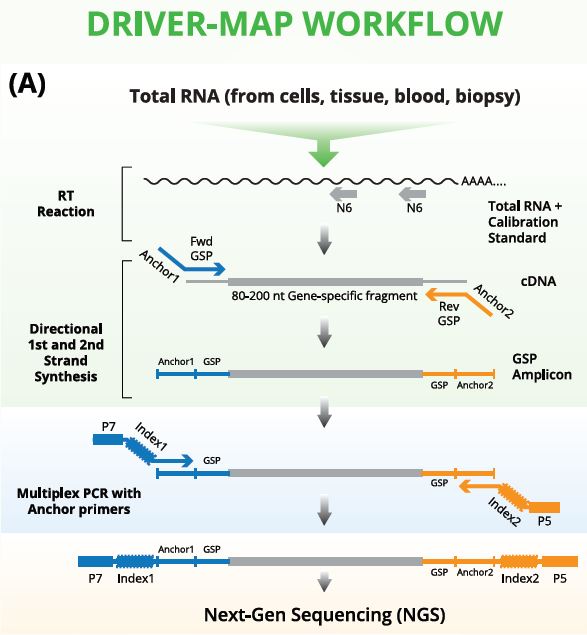
The DriverMap workfiow leverages the power of quantitative PCR with NGS. Experimentally-validated primers amplify specific fixed-length regions of all protein-coding genes in a multiplex reaction. The number of reads of each of the resulting amplicons, as determined by NGS, provides a highly quantitative linear measurement of the abundance of each transcript across a range of 5 orders of magnitude (Panel BJ. Defined amplicons also greatly facilitate alignment and downstream analysis.
The DriverMap Assay uses intelligently designed, empirically optimized targeted primers to amplify defined regions of each transcript for all human genes in a single multiplex RT-PCR reaction. The amplified products of this reaction are then analyzed using Next-Generation Sequencing (NGS) to assess abundance levels.
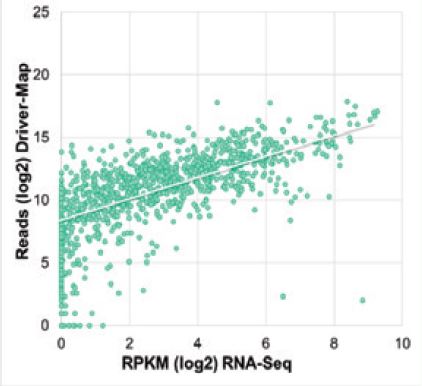
This combination produces an assay that provides the sensitivity of RT-PCR with the dynamic range and quantitation of deep sequencing. Cellecta's novel approach uses total RNA as starting material and provides increased sensitivity for low- abundance genes and a broader linear range for more quantitative differential analysis than RNA-Seq.

A panel of 25 candidate pain biomarkers differentially expressed in individuals with fibromyalgia (FM) vs. healthy control cases. We used genome-wide DriverMap to generate expression data from 50 ng of RNA isolated from whole blood from each of 6 negative controls (no pain) and 7 FM clinical specimens.
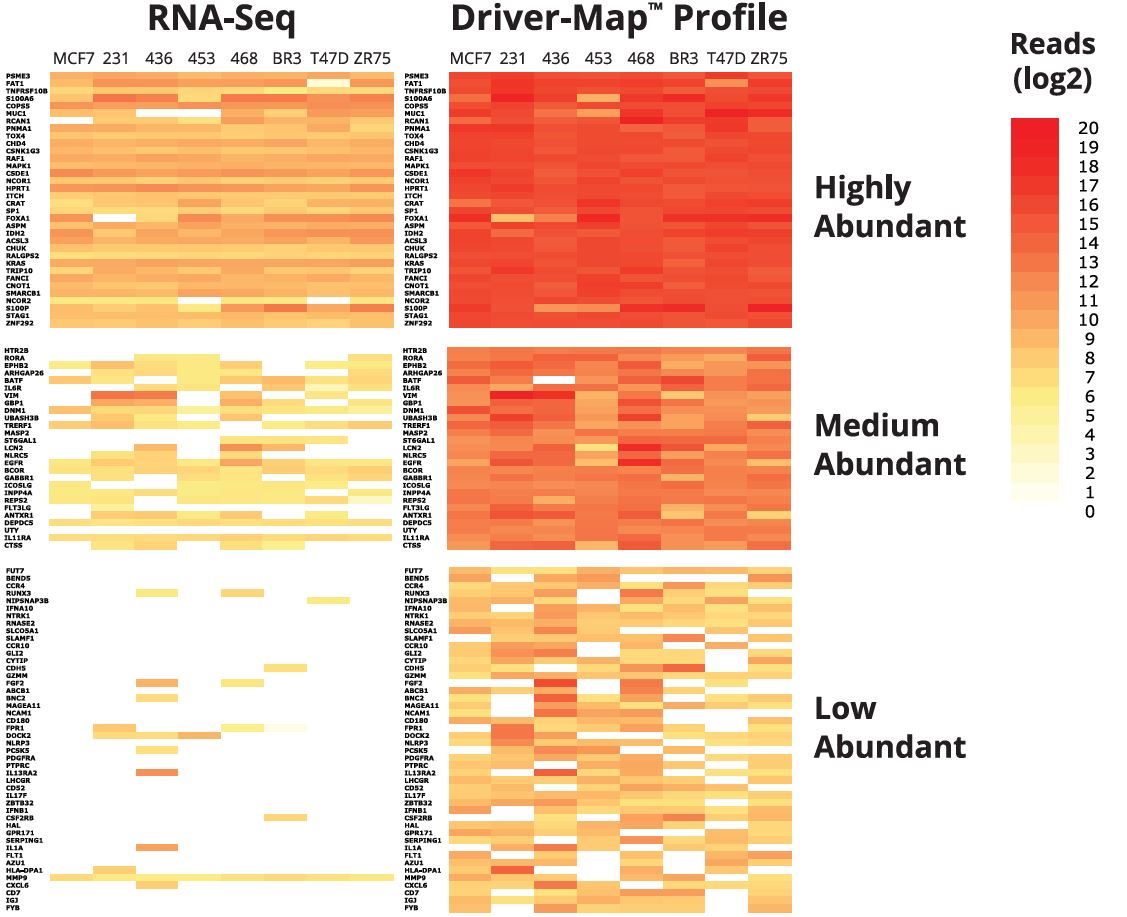
NGS read levels detected RNA-Seq and DriverMap for selected high-abundant (1 0K-1 00K copies per sample), medium-abundant (1,000-10,000 copies per sample), and low-abundant transcripts (100-1,000 copies per sample) in song of total RNA from seven common cancer cell lines.